
細胞培養全般における基本的な工程
公開:2020.01.20
細胞培養の手順や留意事項は、細胞の種類によって異なります。各工程で、各細胞に応じた取り扱いをしなければ、細胞の特性が変化してしまうことがあるため注意が必要です。
ここでは、一般的な細胞の培養工程と注意点を紹介します。
細胞培養に用いる細胞の準備
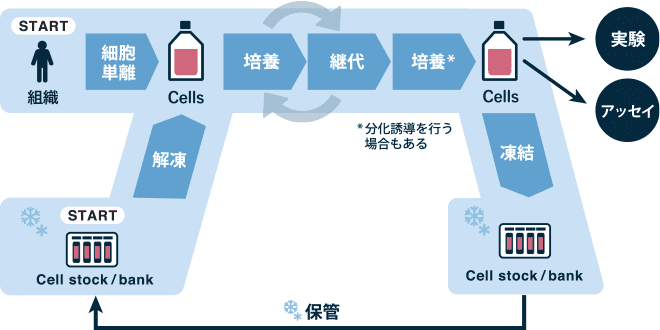
図:細胞の活用の流れ
細胞の入手方法には、細胞バンクから入手する場合と、ドナーから採取した組織から細胞を単離する場合があり、それぞれで培養手順が異なります。
細胞バンクから入手した細胞から培養を開始する場合、「解凍」、「細胞播種」および「細胞観察」の操作を行います。
ドナーから採取した組織では、不要な組織が付着している場合はそれを切除した上で、組織をタンパク分解酵素液に浸漬することなどで細胞を単離します。酵素を用いた場合には、酵素を希釈したり、酵素反応阻害剤により酵素反応を停止した後、「細胞播種」と「細胞観察」の工程を進め、培養する細胞を準備します。
解凍
凍結された細胞を解凍して培養を開始することを、「細胞を起こす」と言います。バンクから入手した細胞の凍結バイアルを液体窒素タンク*1や超低温ディープフリーザー(-150℃)から、液体窒素容器などの適切な保冷容器に入れてベンチまで運び、37℃の恒温水槽または融解装置で融解した後*2、素早く培地を加えて凍結液を薄め、遠心により沈殿させ、凍結液を含む上澄みを除去した後に37℃に加温した新しい培地を添加します。その後ピペッティングにより細胞を懸濁し、顕微鏡や細胞数計測器等を用いて細胞数/細胞濃度を計測します。
*1:液体窒素タンク内での凍結保管方法には、液体窒素の冷気で保管する「気相保存」と、凍結保存するものを直接液体窒素に浸漬する「液相保存」があります。液相保存は、液体窒素の温度である-196℃での保存が可能ですが、凍結バイアル内に液体窒素が入り込む可能性が高く、他のバイアルからの細菌や酵母、マイコプラズマやウイルスのコンタミネーションのリスクが高くなります。*2:液相保存されたバイアルに対しては、液体窒素がバイアルに残っている状態で37℃の恒温水槽または融解装置に入れると、バイアルが破裂する危険があるため、ベンチで一度バイアルの蓋を緩めて締め直してから、37℃の恒温槽に入れます。
細胞播種
目的の細胞播種濃度にするために、計測した細胞数/細胞濃度をもとに、適切な量の培地を加えて希釈し、細胞懸濁液を調整します。ピペッティングにより細胞を均一に懸濁した後、新しい培養容器に所定量の細胞懸濁液を移動します。これを「細胞播種」、また、「細胞を蒔く」と言います。
細胞観察
細胞を新しい培養容器に播種した後、容器の細胞を光学顕微鏡、またはその他の観察装置で以下の通り観察します。
- 細胞の形態を確認して、生存している細胞が多いことを確認する
- 細胞の分布をみて、細胞がまんべんなく位置していることを確認する
- 細胞以外の異物の混入がないか確認する
以上を確認したら、細胞が入っている容器を37℃で加湿したCO2インキュベータに入れて、培養を開始します。
細胞培養開始~継代
細胞観察
新しい培養容器に播種した細胞をCO2インキュベータの中で静置し、一般的には翌日※、光学顕微鏡、またはその他の観察装置で以下の観察作業を行います。
- 細胞の形態や状態を確認することで培養が正常に進んでいるかどうかを判断する
- 細胞以外の異物の混入がないか確認する
培地交換
細胞を解凍した後、CO2インキュベータの中で細胞は増えていきます。細胞は、培養液中から、必要な栄養素を使い、それらの栄養素を代謝します。そのため、栄養分が低下し、代謝産物が多くなった培地を捨てて、新しい培地に交換します。この作業を「培地交換」と言います。
培地交換の前に、まず細胞観察を行い、培養が正常に進んでいるかどうかを確認します。古い培地を捨てた後は、細胞が乾かないように素早く新しい培地を加えます。細胞によっては、培地に浸っていないと死んでしまう場合もあるので、古い培地はすべて捨てないで少しだけ残しておく場合もあります。また、新しい培地は、事前に37℃に温めて、細胞に急激な温度変化を与えないようにします。
培地を交換する作業を行ったら、その作業によるダメージがないか確認するため、細胞観察を行います。また、うまく培養が進んでいることを記録するため、顕微鏡画像を取得して、保存します。
これらの作業が終わったら、再び、インキュベータにて入れて静置し、培養を継続します。
継代
細胞が増えてきたら、培養容器いっぱいになる前に、新しい培養容器に植え替えをします。これを「継代」と言います。培養容器いっぱいに細胞が増えた状態を「コンフルエント」と言います。一般的には、容器の中に占める細胞の面積(占有面積)が、容器いっぱいの手前の70~80%ぐらいの状態で、細胞で継代するのが良いとされています。70~80%ぐらいの状態を「サブコンフルエント」と言います。コンフルエントになってしまうと、細胞と細胞が接触して、「これ以上増えなくていいよ」、と感じてしまい、継代後に増えなくなってしまいます。がん細胞の場合には、それでもどんどん増えてしまいますが、やはり栄養が足りなくなってしまいますので、コンフルエントにすることはよくありません。そのため、細胞がどのくらい増えたか、細胞観察して、モニタリングをしておく必要があります。
継代の方法は、浮遊細胞と接着細胞の場合とで異なります。
浮遊細胞の継代
容器中の細胞を含む培養液をチューブに回収して、遠心して細胞を集めます。沈査を残して、上清を除き、新しい培地に再浮遊させます。その一部を取って、トリパンブルーなどで染色して、生きている細胞数をカウントします。細胞濃度を計算して、適切な細胞密度になるように、細胞希釈方法を考えて、細胞懸濁液の濃度を調整し、新しい容器に入れます。
接着細胞の継代
容器に接着した細胞を、なんらかの方法で容器表面から剥がす必要があります。一般的には、コラゲナーゼ、ディスパーセ、トリプシンなどのプロテアーゼが使用されます。トリプシンの場合には、カルシウムイオンやマグネシウムイオンにより活性が阻害されるため、作用させる前に、これらのイオンを含む培地を洗い流す必要があります。逆に、コラゲナーゼ、ディスパーセなどは、カルシウムイオン存在下で活性を示します。
具体的な手順としては、
- 上清の古い培地を取り除く
- 必要に応じて、リン酸緩衝液、あるいは、培地で洗浄する
- 細胞分散酵素液を添加する
- 酵素の活性温度域内の所定温度で、所定時間、静置し、酵素反応を促進させる
- 細胞の剥離具合を顕微鏡などにて確認する
- タッピング等により細胞の剥離を促進させる
- 酵素反応停止液がある場合には、添加し、酵素反応を停止する。ない場合には、培地を添加して、酵素を希釈して、活性を弱める
- 培地を回収して、遠心により細胞を沈殿させ、酵素と反応停止液が含まれる上清を除去する
- 新しい培地を回収した細胞に添加する
- ピペッティングにより細胞を懸濁し、顕微鏡や重量計測器等を用いて細胞数/細胞濃度を計測する
- 目的の細胞密度となるように調整方法を考えて、培地を添加した後、再度懸濁する
- 新しい培養容器に所定量の細胞懸濁液を播種する
- 顕微鏡観察を行う
- 37℃に加湿したCO2インキュベータに移動する
しかし、細胞によっては、分散酵素を使うと、細胞が弱ってしまう場合があります。そのような場合には、スクレーパーなどでスクレープする、カットする、あるいは、ピペッティングによる水流で剥離などして、機械的に回収します。
細胞培養後の処理(ストックの作製)
培養した細胞は、そのまま使うのではなく、できるだけ早く入手した細胞と同等の形質を持つ状態で、ストックを作っておくことが大事です。細胞は、生きた素材であるため、長期にわたり継代を続けていると、形質が変化して、元の細胞とは異なってしまう場合があるからです。
具体的には、以下の作業で細胞のストックを作製します。
細胞観察
光学顕微鏡、またはその他の観察装置で以下の観察作業を行います。
- 細胞の形態や状態を確認することで培養が正常に進んでいるかどうかを判断する
- 細胞がサブコンフルエントの状態で、増えすぎていないかを判断する
- 細胞以外の異物の混入がないか確認する
関連リンク
通常は目視で行われている、培養中の細胞の増殖能を画像解析により定量評価する事例はこちら
MSCの継代タイミングのばらつき軽減のためにコンフルエント率を数値化通常は培養中の細胞を剥離して行う細胞数カウントを、剥離することなく画像解析により定量評価する事例はこちら
MSCの細胞数と増殖率を非剥離で迅速かつ正確に測定細胞剥離
継代の時と同様に、細胞を回収します。
細胞観察
遠心して回収した細胞に新しい培地を添加しますが、その細胞懸濁液の濃度は、継代の場合よりも濃い細胞濃度にする必要があるため、少量の培地を使用します。これをピペッティングにより細胞を懸濁し、顕微鏡や計測器等を用いて細胞数/細胞濃度を計測します。
分注
目的の細胞密度になるように凍結保存液を添加し、ピペッティングにより再度懸濁します。ピペッティングまたは常時撹拌しながら、所定量ずつクライオチューブに分注します。
凍結
クライオチューブは、速やかに凍結専用容器に入れてディープフリーザーに入れます。あるいは、凍結装置(プログラムフリーザー)にて凍結します。凍結したクライオチューブは保管用液体窒素タンクまたは超低温ディープフリーザー(-150℃)に保管します。
確認
凍結した細胞を一部解凍して培養し、同様に細胞が増え、そのほか、形質が同様であることを確認します。それが確認できたら、最初に培養をしていた細胞も、同様に凍結をします。
実験開始
ストックとして作製した細胞を解凍して、研究などに使用します。一定期間以上培養を継続したら、その細胞は廃棄し、また、ストックを解凍して、使用します。

